CASE REPORT | https://doi.org/10.5005/jp-journals-10055-0091 |
Optic Nerve Enlargement in Krabbe’s Disease: A Case Report
1,2Department of Radiodiagnosis and Imaging, Sri Guru Ram Das Institute of Medical Sciences and Research, Amritsar, Punjab, India
Corresponding Author: Vijinder Arora, Department of Radiodiagnosis and Imaging, Sri Guru Ram Das Institute of Medical Sciences and Research, Amritsar, Punjab, India, Phone: +91 7973561408, e-mail: drvijinderarora338@gmail.com
How to cite this article Arora V, Goindi AS. Optic Nerve Enlargement in Krabbe’s Disease: A Case Report. AMEI’s Curr Trends Diagn Treat 2020;4(1):33–35.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Krabbe’s disease is an autosomal recessive leukodystrophy that presents clinically with increased body tone, regression of milestones, excessive irritability, and inconsolable crying. The pathologic basis of the disease is abnormal myelin metabolism resulting from a deficiency in the galactosylceramidase enzyme with subsequent white matter destruction. Infantile form is the most common subtype, occurring at about 6 months of age. Here, we report a case of Krabbe’s disease in which there was associated enlargement of the intracranial optic nerves. The purpose is to acknowledge various imaging features of Krabbe’s disease on computed tomography (CT) scan and magnetic resonance imaging (MRI) and to have Krabbe’s disease included as one of the differential diagnosis in children with enlargement of the optic nerves. Imaging findings with Krabbe’s disease include areas of increased attenuation in the region of thalamus on CT scans, areas of abnormal signal in the white matter on MR images, and white matter volume loss. Hypertrophy of optic nerves is an unusual finding rarely seen in Krabbe’s disease. The diagnosis of Krabbe’s disease is challenging because of the large number of demyelinating diseases and leukodystrophies as well as the similarities between their clinical and imaging findings. Specific neuroimaging findings and clinicobiochemical correlation may help to establish this diagnosis.
Keywords: Demyelination, Hypertrophy, Krabbe’s disease, Leukodystrophy, Optic nerve.
INTRODUCTION
Krabbe’s disease is a rare type of lysosomal storage disorder. It shows autosomal recessive inheritance and occurs due to abnormal enzymatic activity of galactosylceramidase resulting in accumulation of galactosylceramide and galactosylsphingosine. It has an approximate incidence of 1 case for 100,000–200,000 live births.1,2
Infantile form is the most common subtype that usually occurs around 6 months of age and clinically presents with neurodegenerative symptoms, progressive spastic quadriparesis, tonic spasms, inconsolable crying, and extreme irritability. Secondary blindness may occasionally occur due to progressive optic atrophy.3
CASE DESCRIPTION
A six and a half months old male infant presented with symptoms of pronounced irritability in the form of relentless crying and random episodes of stiffening of the body since birth. No developmental delay was found at the time of scan. There was no significant birth or family history provided by the attendants. A complete clinical examination showed that the patient was very irritable and had increased tone in both truncal and appendicular regions. Computed tomography (CT) scan of the brain depicted areas of hyperdensities involving bilateral thalami (Fig. 1). Magnetic resonance imaging (MRI) brain revealed areas of abnormal hyperintensities on T2-weighted (T2W) images in the periventricular white matter (Fig. 2). The optic chiasma and bilateral optic nerves in the prechiasmatic region were uniformly enlarged. However, the intraorbital part of both optic nerves appeared normal in thicknes (Fig. 3). A diagnosis of Krabbe’s disease was made having characteristic imaging findings on CT scan and MRI. Biochemical confirmation of the diagnosis could not be made, as the patient did not go in for further investigation.
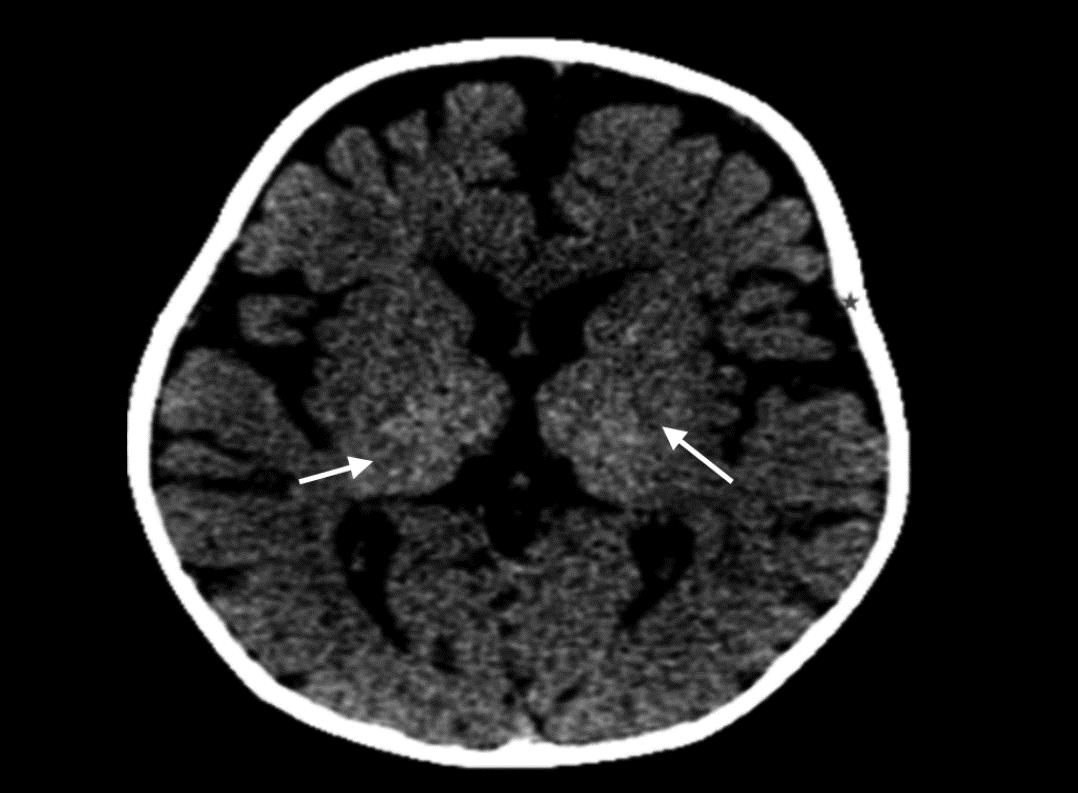
Fig. 1: CT scan showing areas of hyperdensities involving bilateral thalami (arrows) with diffuse cerebral atrophy
DISCUSSION
Krabbe’s disease is an inherited lysosomal storage disorder with characteristic imaging features on both CT scan and MRI. On CT scan, diffuse hyperdense areas depicting high attenuation are seen in the bilateral thalami, caudate nuclei, and corona radiata. Diffuse white matter atrophy occurs as the disease progresses. On MRI, scattered areas of abnormal T2 hyperintensities are seen. Features of global brain atrophy in the form of prominence of cerebral and cerebellar folia appear in the later stages of the disease.3
In our patient, the typical neuroimaging features of Krabbe’s disease were associated with hypertrophy of the prechiasmatic intracranial segment of the bilateral optic nerves, which is an unusual associated finding.4
Krabbe’s disease usually presents within the first 6 months after birth. Most common clinical features include stiffness, increased tone of the body, fever, hyperirritability regression of milestones, and developmental delay. A further decline in cognition with features such as opisthotonos, myoclonus, nystagmus and atrophy of optic nerves occur in the later stages. Symptoms eventually lead to spastic paralysis with the lack of reaction to peripheral stimulus and absence of spontaneous motion along with marked involvement of the peripheral nerves eventually causing hypotonia rather than irritability. Involvement during childhood or adulthood may be seen less frequently.5–7
A faulty gene on chromosome 14 forms is responsible for pathogenesis of Krabbe’s disease.8 It is a rare autosomal recessive lysosomal storage disorder. The enzymatic abnormalities result in defective lysosomal function that results in deposition of galactosylsphingosine (psychosine) in the brain. The amount of accumulated galactosylsphingosine in the diseased brain tissue can be upto 100 times more the usual levels.9 Usually, galactosylceramidase I (galactocerebroside beta-galactosidase) enzyme helps in removing galactose from galactosylceramide and galactosylsphingosine leaving behind ceramide and sphingosine, respectively. In Krabbe’s disease, the Galactosylceramidase I enzyme deficiency occurs. Galactosylceramidase II and III enzymes can help in the cleavage of galactosylceramide but not galactosylsphingosine. Galactosylsphingosine is harmful for the brain cells and frequently targets oligodendroglial cells and hampers the myelin formation. However, the exact mechanism of dysmyelination and demyelination is still not clear.6
Main histopathologic findings of Krabbe’s disease include marked depletion of oligodendroglial cells and the appearance of globoid cells. The latter signifies high quantity of galactocerebroside within the macrophages. Oligodendrocyte loss occurs due to increased galactosylsphingosine levels, while globoid cells are formed as a result of the increased accumulation of galactosylceramide within the cells. Sulfotransferase is another enzyme that may be depleted in Krabbe’s disease, indicating a more complex mechanism of galactosylceramide degradation by these enzymes.7
Although the diffuse atrophy appears to be an inconsistency with findings like enlarged optic nerves seen in cases of Krabbe’s disease, it may represent of the two major histologic varities of the enzyme deficiency. The global atrophy may occur due to loss of oligodendrocytes from galactosylsphingosine toxicity, and the increased bulk of optic nerves occurs due to the accumulation of globoid cells. However, the reason for the isolated hypertrophy of optic nerves remains unclear.3
Optic nerve enlargement has multiple differentials in the pediatric age-group such as dural ectasia, nerve sheath meningioma, optic nerve glioma, and histiocytic/granulomatous involvement of the optic nerves. Other clinical conditions such as orbital pseudotumor, leukemia, postviral optic nerve inflammation, juvenile xanthogranuloma, optic nerve medulloepithelioma, and invasion of the optic nerve/infiltration of the nerve head with retinoblastoma may also be included in the differential diagnosis.3,10 Ectasia of the optic nerve sheath and optic nerve glioma also occur in neurocutaneous syndromes such as neurofibromatosis-1 (NF-1). As NF-1 is a much more commonly seen entity than Krabbe’s disease, the combination of features like optic nerve enlargement and patchy white matter signal abnormalities on T2W images would logically lead to NF-1 as the diagnosis by imaging criteria. However, the presenting features of both these entities are significantly different, where children having Krabbe’s disease show severe hyperirritability and altered muscular tone in contrast to children with NF-1 who present with seizures, developmental delay with some characteristic skin lesions.3
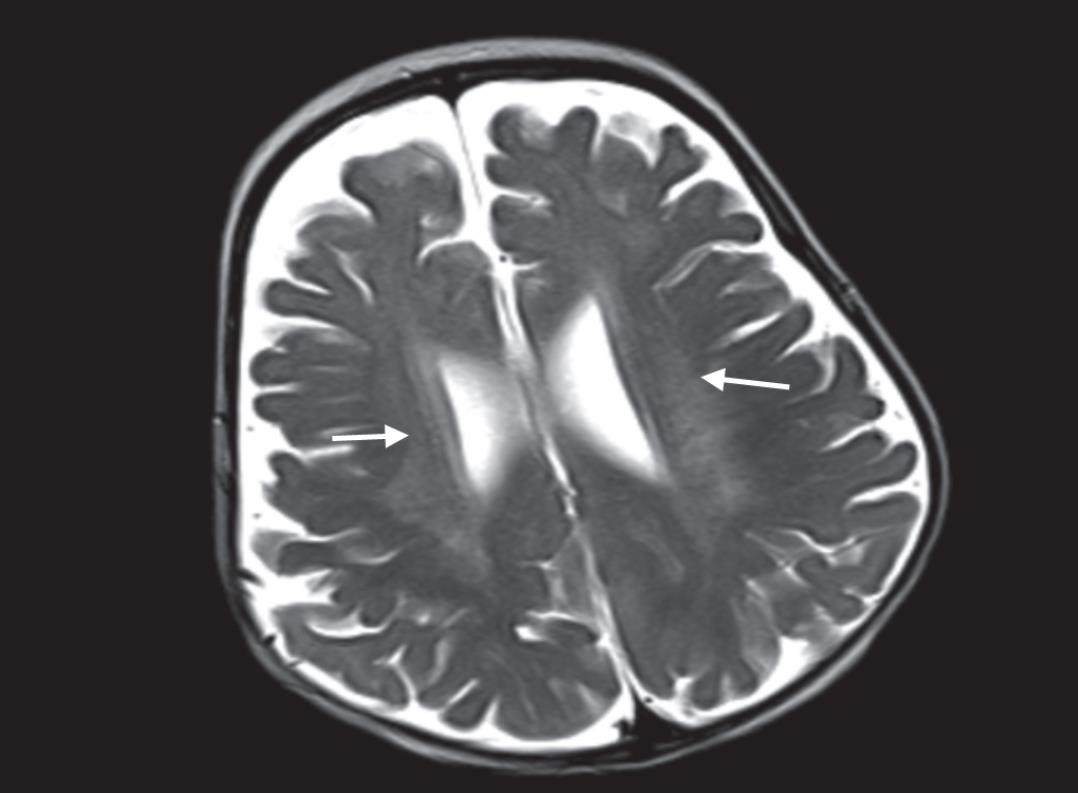
Fig. 2: Areas of abnormal signal seen in periventricular white matter on axial T2W image

Figs 3A to D: (A) Axial T1W MRI image showing thickened optic nerves at prechiasmatic segment; (B) Axial T2W MRI image showing thickened optic nerves; (C) Optic nerve hypertrophy on T2W sagittal image; (D) Optic nerve hypertrophy on coronal T2W image
REFERENCES
1. Kamate M, Hattiholi V. Normal neuroimaging in early-onset Krabbe disease. Pediatr Neuro 2011;44(5):374–376. DOI: 10.1016/j.pediatrneurol.2010.12.008.
2. Bussière M, Kotylak T, Naik K, et al. Optic nerve enlargement associated with globoid cell leukodystrophy. Can J Neurol Sci 2006;33(2):235–236. DOI: 10.1017/S0317167100005047.
3. Jones BV, Barron TF, Towfighi J. Optic nerve enlargement in Krabbe’s disease. AJNR Am J Neuroradiol 1999;20(7):1228–1231.
4. Castilha-Neto D, Monteiro LF, Peruchi MM, et al. Optic nerve enlargement in infantile form of Krabbe disease. Clin Pract 2012;2(4):81. DOI: 10.4081/cp.2012.e81.
5. Kwan E, Drace J, Enzmann D. Specific CT findings in Krabbe’s disease. AJNR Am J Neuroradiol 1984;5:453–458.
6. Volpe JJ. Hypoxic ischemic encephalopathy. Neurology of the newborn.Philadelphia: W.B. Saunders Company; 1995. pp. 573–574.
7. Ruggieri PM. Metabolic and neurodegenerative disorders and disorders with abnormal myelination. In: ed. WS, Jr Ball ed. Pediatric Neuroradiology. Philadelphia: Lippincontt-Raven; 1997. pp. 194–197.
8. Zlotogora J, Chakraborty S, Knowlton RG, et al. Krabbe’s disease locus mapped to chromosome 14 by genetic linkage. Am J Hum Genet 1990;47(1):37–44.
9. Baram TZ, Goldman AM, Percy AK. Krabbe’s disease: specific MRI and CT findings. Neurology 1986;36(1):111–115. DOI: 10.1212/WNL.36.1.111.
10. Garcia AML, Morais NMM, Ohlweiler L. Optic nerve enlargement and leukodystrophy. Arq Neuropsiquiatr 2010;68(5):816–818. DOI: 10.1590/S0004-282X2010000500029.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.